Chronic lymphocytic
leukemia (CLL) was largely considered to be a disease of slow progression,
standard treatment with Chlorambucil and having almost similar prognosis. With
the introduction of molecular methods for understanding the disease
pathophysiology in CLL there has been a remarkable change in the approach
towards the disease. The variation in B-cell receptor response and immunoglobulin
heavy chain variable region (IGHV) mutation, genetic aberration and defect in
apoptosis and proliferation has had an impact on therapy initiation and
prognosis. Early diagnosis of molecular variant is therefore necessary in CLL.
Chronic lymphocytic leukemia is a type of cancer in which the bone
marrow makes too many lymphocytes (a type of white blood cell).
Chronic
lymphocytic leukemia (also called CLL) is a blood and bone
marrow disease that
usually gets worse slowly. CLL is the second most common type of leukemia in adults. It often occurs during or
after middle age; it rarely occurs in children.
Normally, the body makes blood stem
cells (immature cells) that become mature blood cells over time. A
blood stem cell may become a myeloid stem cell or a lymphoid stem cell.
A myeloid stem cell becomes one of three types
of mature blood cells:
·
Red blood cells that
carry oxygen and other substances to all tissues of the body.
·
White
blood cells that fight infection and disease.
·
Platelets that form blood
clots to stop
bleeding.
A lymphoid stem cell becomes a lymphoblast cell and then one of three types
of lymphocytes (white blood cells):
·
B lymphocytes that
make antibodies to help fight infection.
·
T
lymphocytes that help B lymphocytes
make antibodies to fight infection.
·
Natural
killer cells that
attack cancer cells and viruses.
Blood cell
development. A blood stem cell goes through several steps to become a red blood
cell, platelet, or white blood cell.
In CLL, too many blood stem cells become abnormal lymphocytes and do not become healthy
white blood cells. The abnormal lymphocytes may also be called leukemia cells.
The lymphocytes are not able to fight infection very well. Also, as the number
of lymphocytes increases in the blood and bone marrow, there is less room for
healthy white blood cells, red blood cells, and platelets. This may cause
infection, anemia, and easy bleeding.
This summary is about chronic lymphocytic
leukemia. See the following PDQ summaries for more information about
leukemia:
Older age can affect the risk of developing chronic lymphocytic
leukemia.
Anything that increases your risk of getting a
disease is called a risk
factor. Having a risk factor
does not mean that you will get cancer; not having risk factors doesn’t mean
that you will not get cancer. Talk with your doctor if you think you may be at
risk. Risk factors for CLL include the following:
·
Being middle-aged or
older, male, or white.
·
A family
history of CLL or cancer
of the lymph
system.
·
Having relatives who
are Russian Jews or Eastern European Jews.
Possible signs of
chronic lymphocytic leukemia include swollen lymph nodes and tiredness.
Usually CLL does not cause any symptoms and is found during a routine blood
test. Sometimes symptoms
occur that may be caused by CLL or by other conditions. Check with your doctor if you have any of
the following problems:
·
Painless swelling of
the lymph
nodes in the neck,
underarm, stomach, or groin.
·
Feeling very tired.
·
Pain or fullness below
the ribs.
·
Fever and infection.
·
Weight loss for no
known reason.
Tests that examine the
blood, bone marrow, and lymph nodes are used to detect (find) and diagnose
chronic lymphocytic leukemia.
The following tests and procedures may be
used:
·
Physical exam and history : An exam of the body to check
general signs of health, including checking for signs
of disease, such as lumps or anything else that seems unusual. A history of the
patient’s health habits and past illnesses and treatments will also be taken.
·
Complete
blood count (CBC) with differential : A procedure in which a sample of blood is drawn and
checked for the following:
o The number of red blood cells and platelets.
o The number and type of white blood cells.
o The amount of hemoglobin (the protein that carries oxygen) in the red blood
cells.
o The portion of the blood sample made up of red
blood cells.
Complete blood count (CBC). Blood is collected
by inserting a needle into a vein and allowing the blood to flow into a tube.
The blood sample is sent to the laboratory and the red blood cells, white blood
cells, and platelets are counted. The CBC is used to test for, diagnose, and monitor
many different conditions.
·
Immunophenotyping : A laboratory
test in which
the antigens or markers on the surface of a blood or bone marrow
cell are checked to see if they are lymphocytes or myeloid cells. If the cells
are malignant lymphocytes (cancer), they are checked
to see if they are B lymphocytes or T lymphocytes.
·
FISH (fluorescence in situ hybridization): A laboratory technique used to look at genes orchromosomes in cells and tissues. Pieces of DNA that contain a fluorescent dye are made
in the laboratory and added to cells or tissues on a glass slide. When these
pieces of DNA bind to specific genes or areas of chromosomes on the slide, they
light up when viewed under a microscope with a special light.
·
Flow
cytometry : A laboratory
test that measures
the number of cells in a sample, the percentage of live cells in a sample, and
certain characteristics of cells, such as size, shape, and the presence
of tumor
markers on the cell
surface. The cells are stained with a light-sensitive dye, placed in afluid, and passed in a stream before a laser or other type of light. The measurements
are based on how the light-sensitive dye reacts to the light.
·
IgVH gene mutation test: A laboratory test done on a bone marrow or blood sample to
check for an IgVH gene mutation. Patients with an IgVH gene
mutation have a better prognosis.
·
Bone
marrow aspiration and biopsy : The removal of bone marrow, blood, and
a small piece of bone by inserting a hollow needle into the hipbone or breastbone. A pathologist views the bone marrow, blood, and bone
under a microscope to look for abnormal cells.
Bone marrow aspiration and biopsy. After a
small area of skin is numbed, a Jamshidi needle (a long, hollow needle) is
inserted into the patient’s hip bone. Samples of blood, bone, and bone marrow
are removed for examination under a microscope.
Certain factors affect
treatment options and prognosis (chance of recovery).
Treatment options depend on:
·
The stage of the disease.
·
Red blood cell, white
blood cell, and platelet blood counts.
·
Whether there are
symptoms, such as fever, chills, or weight loss.
·
Whether the liver, spleen, or lymph nodes are larger than normal.
·
The response to initial treatment.
·
Whether the CLL
has recurred (come back).
The prognosis (chance of recovery) depends on:
·
Whether there is a
change in the DNA and the type of change, if there is one.
·
Whether lymphocytes
are spread throughout the bone marrow.
·
The stage of the
disease.
·
Whether the CLL gets
better with treatment or has recurred (come back).
·
Whether the CLL progresses to lymphoma or prolymphocytic leukemia.
·
The patient's general
health.
Diagnosis
CLL can be diagnosed if: the clonal B-lymphocyte
count is >5,000/cuml of blood (duration of lymphocytosis >2 months). If
the lymphocyte count is less than 5,000/cuml in asymptomatic individuals,
without organ involvement, it is designated as 'monoclonal B lymphocytosis'.
-Bone marrow lymphocytes >30%
-The immunological profile of CLL lymphocytes is
defined by:
- Weak
surface membrane immunoglobulin (Ig) levels (most often IgM or both IgM
and IgD)
- Monoclonal:
expression of either Kappa or lambda
- B-cell
antigens CD23, CD19 and CD20 (weak), with co-expression of CD5
- Negative
for cyclin D1 and CD10 expression,
No or
weak expression of FMC7, CD22 and CD79b.
Differential diagnosis of CLL includes
-Mantle cell Lymphoma
-Follicular lymphoma
-Splenic marginal zone lymphoma
-Hairy cell lymphoma
-B Prolymphocytic Leukemia.
Staging at diagnosis (Rai system)
0. Lymphocytosis
1. Lymph node enlargement
2. Spleen enlargement
3. Hemoglobin < 11 g/dl
4. Platelets < 100,000/μl
Staging at diagnosis (Binet system)
- Lymphocytosis
- Lymph
node enlargement in > 3 areas
- Cytopenia
: h0 emoglobin < 10 g/dl or platelets <100,000/μl
Pathophysiology of chronic lymphocytic
leukemia
Most of the CLL cells are inert in vitro. Recently, the understanding of the patho-biology in CLL has divided this
disease into subgroups and this has had a profound impact on prognosis and
treatment.
The three major subdivisions are based upon:
1. B-cell
receptor response and the IGHV mutation,
2. Genetic
aberration/gene mutation,
3. Defects
in apoptosis and proliferation.
B-cell receptor response
The B-cell receptor (BCR) is composed of two
immunoglobulins (Ig), heavy and light chains (variable and constant region) and
CD79a and CD79b. These contain intracellular activation molecules (SYK and LYN)
that transmit signals to intracellular tyrosine kinases [Figure 2]. The
ability of these kinases to activate downstream pathways varies in CLL
subgroups and is correlated with:
- Ig
heavy chain variable region (IGHV) mutational status,
- ZAP-70
and
- CD
38 expression.
These
pathways can be targeted by small molecule inhibitors, the most promising of
which might be SYK inhibitors.
B-cell receptor response and the IGHV mutation
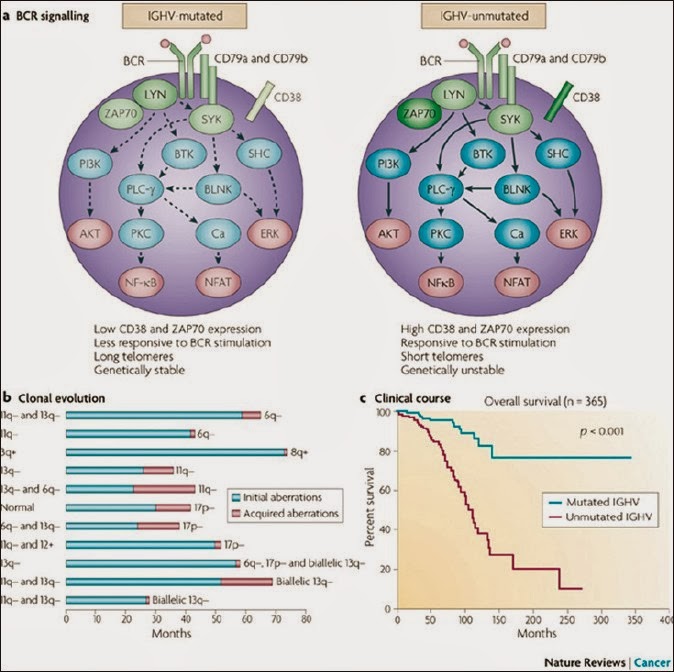
The B cell response to antigenic stimulation is
mediated through the BCR in normal and malignant B-cells.
Each B-cell displays a distinct BCR that is
formed through variable combinations of V, D and J segments for the Ig heavy
chain and V and J gene segments for the light chain.
CLLs have mutated IGHV genes and unmutated IGHV
genes (unmutated IGHVs showing poorer survival).
(IGHV genes = immunoglobulin heavy chain
variable region)
Folding and glycosylation defect of the μ and
CD79A chains (not of the CD79B chain).
Poor expression of the CD22 molecule in B-cell
chronic lymphocytic leukemia cells was also as a result of folding defect
arising in CD79A.
Most B-cell chronic lymphocytic leukemia cells
express CD5 and IgM/IgD due to which they have a mantle zone-like phenotype of
naive cells that., …, express unmutated immunoglobulin genes normally. Somatic mutations of IGHV genes are seen in Fifty to
seventy percent of cases of chronic lymphocytic leukemia., appearing as if they had matured from a lymphoid
follicle.
Mutational status of IGHV genes has a profound
effect on the prognosis of CLL showing markedly different biological and
clinical behaviors. BCR surface expression is usually weak in CLL. Low
expression of the B-cell receptor is the typical presentation of lymphocytes in
CLL (mechanisms remain elusive).
ZAP70 (Zeta-Associated Protein)
Low expression of the B-cell receptor correlates
with defective intracellular calcium mobilization and tyrosine phosphorylation
due to reduced induction of protein tyrosine kinase activity. High protein tyrosine kinase activity associated with
ZAP70-a receptor (not found in normal circulating B-cells) is usually found in
T-cells and natural killer cells detected in most patients with un-mutated
chronic lymphocytic leukemia.
ZAP70 expression is associated with advantageous
survival of CLL cells because of enhanced access to proliferation centre and
increased sensitivity to chemokine migratory signals (presence of ZAP70 is an oncogenic event in CLL,
concentrated particularly in lymph node lymphoid cell).
CD38
CD38, which is associated with poor prognosis,
predominates in patients with unmutated IGHV genes.
In chronic lymphocytic leukemia, expression of
CD38 on B cells favors -. its growth and survival through sequential
interactions between CD38 and CD31 and also between CD100 and plexin B1
(PLXNB1).
Genetic abnormalities
Dohner and colleagues .- in a series of 325 patients with chronic
lymphocytic leukemia showed that chromosomal aberrations can be detected in
interphase cells by fluorescence in
situ hybridization (FISH)
technique in 82% of cases. According to them the most frequent aberrations are:
- Deletion
on Chromosome 13q (55%),Trisomy 12 (18%), A deletion on Chromosome 11q
(16%)., Deletion on chromosome 17p, affecting the TP53 protein, is seen
less frequently (7%).
Deletions in band 13q14
'Deletion on Chromosome 13q' is the most
frequently found genetic structural aberration in CLL and confers a favorable
course. No inactivation of candidate genes by mutation has
been demonstrated. Two microRNA genes, mir-15a and mir-16-1, located in the
crucial 13q14 region have been implicated in CLL pathogenesis. A mouse model with a targeted
deletion of the mir-15a-mir-16-1 locus recapitulates many features of CLL. This
suggests that miR-15a and miR-16-1 have a direct pathogenetic role in CLL.
Deletions at 13q14 occur at high frequencies in
other lymphomas and solid tumors, and a recent study has implicated miR-15a and
miR-16-1 in the pathogenesis of prostate cancer through their targeting of
cyclin D1 and WNT3A, which promote survival and proliferation.
Deletions of ATM (11q22-q23)
Although they are rarely found in early-stage
disease, approximately one-quarter of patients with advanced CLL have 11q23
deletions. Correspondingly, patients who have 11q23 deletions have a more rapid
disease progression and extensive lymphadenopathy. In stuacminimal consensus region in chromosome bands
11q22.3-q23.1 which also harbors the ataxia telangiectasia-mutated (ATM) gene
in almost all cases.
ATM mutations have been shown to be present in
12% of all patients with CLL and in approximately one-third of the cases with a
11q23 deletion. The ATM protein kinase is a central component of the
DNA damage pathway and mediates cellular responses to DNA double-strand breaks
(DSBs). ATM deficiency leads to ataxia-telangiectasia, which is characterized
by extreme sensitivity to irradiation, genomic instability and a predisposition
to lymphoid malignancies. ATM activates cell cycle checkpoints, can induce
apoptosis in response to DNA breaks and functions directly in the repair of DNA
DSBs by maintaining DNA ends in repair complexes.
Trisomy 12
Trisomy 12 is among the more frequent
aberrations in CLL (10-20%),. the genes implicated in the pathogenesis of CLL
with Trisomy 12 are unknown. A previously described association with poor
outcome has not been confirmed. Incidence of Trisomy 12 does not increase with
advanced stage or progression to refractory disease.
Deletions in band 17p13 or TP53 mutations
Deletion of 17p13 is found in 4-9% of CLLs at
diagnosis or at initiation of the first treatment.17p13 deletion usually encompasses most of the short
arm of Chromosome 17p, the deletion always includes band 17p13, where the tumor
suppressor TP53 (which encodes p53) is located. Among CLL cases that have
monoallelic 17p13 deletions, the majority show mutations in the remaining TP53
allele (>80%). Among cases without 17p13 deletion, TP53 mutations are much
rarer [Figure 4].

TP53 mutations in chronic lymphocytic leukemia
TP53- tumor suppressor gene, is a transcription
factor activated by strand breaks in DNA which can trigger apoptosis or cell
cycle arrest.
The Genetic lesions associated with deletions of
the short arm of Chromosome 17 (del17p13)). encodes the TP53 -. gene. The long
arm of Chromosome 11 (del11q23), which encodes the ataxia telangiectasia
mutated (ATM) gene can also result in a loss of function of TP53.
Patients with a 17p13 deletion or TP53 mutation
have a poor outcome and are candidates for experimental strategies with novel
agents and stem cell transplantation. 17p13 deletion is invariably associated with loss of
TP53 as confirmed by fluorescence in
situ hybridization (FISH) ('+'
in [Figure 4] denotes a TP53 deletion). Multiple genomic aberrations
target the p53 pathway in CLL.
Recurrent translocations
Recurrent translocations are rare in CLL in
contrast to other types of leukemia or B-cell lymphoma in which specific and
recurrent Ig locus-associated translocations deregulate known oncogenes.
However, IGHV-mutated CLLs, in which the precursors were exposed to SHM
(Somatic Hyper Mutation) and, in a fraction of cases, also had undergone class
switching. The lack of Ig-associated translocations may support the notion that
CLL is derived from post-GC B-cells, in which SHM and class switching are
silenced.
Microenvironment
CLL cells interact with and seem to shape their
microenvironment, which consists of T-cells, stromal cells and soluble factors. CLL cells rapidly undergo apoptosis when they are
removed from patients. This process can be prevented by adding a number of
cytokines or other cell types to the CLL cell culture.
In the lymph node, the microenvironment provides
anti-apoptotic signals and proliferative stimuli, resulting in the formation of
proliferation centers of CLL cells (pseudo follicles) that are not found in
other lymphomas.
CLL cells seem to recruit accessory cells and thereby create a
microenvironment that supports their own survival.
There is an increase of CD3+ T-cells, most of
which are CD40L+CD4+, and cluster in and around pseudo follicles. CLL cells that are in close proximity and in contact
with activated CD4+ T-cells show expression of the cell surface marker CD38.
This is of interest because CD38 has been linked to the proliferation of CLL
cells. The presence of high numbers of CD38+ CLL cells in the blood is
associated with a poor prognosis.
High-risk features for chronic lymphocytic
leukemia
1. CD38
expression in > 30% of lymphocytes
2. ZAP70
expression in > 30% of lymphocytes
3. Unmutated
(germ line) IgVH gene
4. High-risk
cytogenetic abnormalities
a. 14q
changes
b. 11q
changes
c. 17p
depletion
d. Trisomy
12
5. Rai Stage
3 or 4 or Binet Stage C
6. Doubling
time of lymphocyte count <12 months
7. Elevated
beta-2 microglobulin
8. Elevated
serum thymidine kinase
Presence
The following stages are
used for chronic lymphocytic leukemia:
In stage
0 chronic lymphocytic leukemia, there are too many lymphocytes in the blood, but there are no other symptoms of leukemia. Stage 0 chronic lymphocytic leukemia
is indolent (slow-growing).
In stage
I chronic lymphocytic leukemia, there are too many lymphocytes in the blood and the lymph
nodes are larger than
normal.
In stage
II chronic lymphocytic leukemia, there are too many lymphocytes in the blood, the liver orspleen is larger than normal, and the lymph
nodes may be larger
than normal.
In stage
III chronic lymphocytic leukemia, there are too many lymphocytes in the blood and there are too few red
blood cells. The lymph
nodes, liver, or spleen may be larger than normal.
In stage
IV chronic lymphocytic leukemia, there are too many lymphocytes in the blood and too fewplatelets. The lymph
nodes, liver, or spleen may be larger than normal and there may
be too few red
blood cells.
After chronic lymphocytic leukemia has been diagnosed, tests are
done to find out how far the cancer has spread in the blood and bone marrow.
Staging is
the process used to find out how far the cancer has spread. It is important to know
the stageof the disease in order to plan the best
treatment. The following tests may be used in the staging process:
·
Chest x-ray : An x-ray of the organs and bones inside the chest. An x-ray is
a type of energy beam that can go through the body and onto film, making a
picture of areas inside the body, such as the lymph
nodes.
·
MRI (magnetic resonance imaging): A procedure that uses a magnet, radio waves,
and a computer to make a series of detailed pictures of areas inside the body,
such as the brain andspinal
cord. This procedure is
also called nuclear magnetic resonance imaging (NMRI).
·
CT
scan (CAT scan): A procedure that makes a series of detailed
pictures of areas inside the body, taken from different angles. The pictures
are made by a computer linked to an x-ray machine. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is
also called computed tomography, computerized tomography, or computerized axial
tomography.
·
Blood
chemistry studies : A procedure in
which a blood sample is checked to measure the amounts
of certain substances released into the blood by organs and tissues in the
body. An unusual (higher or lower than normal) amount of a substance can be
a sign of disease in the organ or tissue that
makes it.
·
Antiglobulin
test : A test in
which a sample of blood is looked at under a microscope to find out if there are any antibodies on the surface of red
blood cells or platelets. These antibodies may react with and destroy
the red blood cells and platelets. This test is also called a Coomb's test.
Genotype-specific
therapy
The first risk-adapted treatment for patients
with CLL has been developed for patients with 17p13 deletions who have a very
poor prognosis with alkylator- and purine analogue-based chemo-immunotherapy.There are evidences that
several 'biological' agents, such as alemtuzumab, corticosteroids, lenalidomide
and flavopiridol act independently of functional p53 in CLL, therefore the
current treatment approaches in clinical trials use these agents before going
for allogeneic stem cell transplantation.
Translating biological insights into
treatment
The clinical course is generally indolent in the
majority of patients and therefore clinical endpoints are reached slowly. In
spite of this, the growing number of approaches that act differently from
classical chemotherapy show great promise and some of them are particularly
promising for CLL (for example, SYK inhibition).
Conclusions and perspectives
CLL can be divided into subtypes (IGHV-unmutated
and mutated) that have distinct biological and clinical characteristics.
IGHV-mutated CLLs derive from post-GC B-cells, and that IGHV-unmutated CLLs
stem from B-cells that have been activated by antigens. The dependence and
interaction of CLL cells with the microenvironment is of pivotal importance and
is being used as a drug target in preliminary studies.
Specific aberrations (11q23 and 17p13 deletions)
and gene mutations (TP53 and ATM) help to define distinct biological and
clinical subgroups of CLL. The most common genetic lesion-deletion 13q14-may
serve as a model of how deregulated non-coding RNAs contribute to cancer
initiation, and this may become a 'druggable' target in the future.
Overall, CLL may serve as a model for how
microenvironmental stimuli, antigenic drive and genetic deregulation are
combined in cancer pathogenesis. Importantly, there are a growing number of
agents that act on specific biological targets and therefore open of
large-cell transformation (Richter's syndrome)
GOT THESE FROM DIFFERENT SITES , I AM DOING MY PROJECT ON IT













